The Basic search accepts diverse entries, including:
- Gene and gene tree identifiers
- gene, transcript or protein names
- Uniprot protein names or accession numbers
- cross-references (Gene Ontology, InterPro, ...)
- keywords
By default, a wildcard is added at the end of each keyword. For an exact match search add +, for example +maltase.
Only words with 3 or more characters are matched. For example, p53 will not only match p53 but also words longer than 3 characters, starting with p53.
Some query examples:
inhibitor
=> inhibitor*
+inhibitor
=> inhibitor
inhibitor protein
=> inhibitor* AND protein*
inhibitor +protein
=> inhibitor* AND protein (without proteinase!)
"inhibitor heavy"
=> "inhibitor heavy" phrase
inhibitor human
=> inhibitor* IN human
inhibitor -human
=> inhibitor* NOT IN human
GO:0042030
=> Gene Ontology term
And combinations...
You can use the Advanced search panel to perform a search on a specific phylogenetic branch. Branch names follow the NCBI Taxonomy.
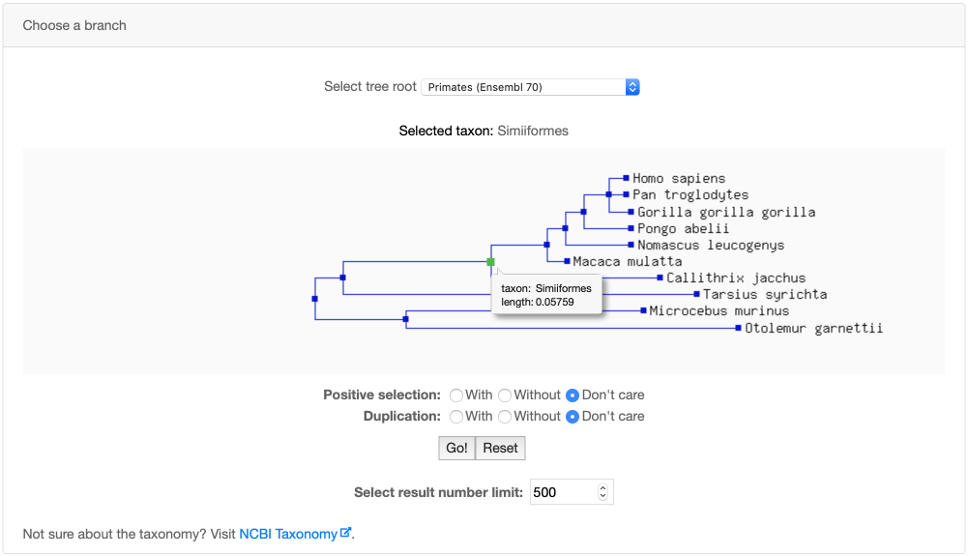
If Positive selection is selected, the query will search only for subtrees with significant positive selection on the chosen branch.
Note that the query is carried out on subtrees. Thus a query for the Simiiformes branch with and without positive selection, may return some common trees
because some subtrees may contain duplicated Simiiformes branches with AND without positive selection events.
Positive selection detection on terminal branches has lower statistical power, and sequencing or gene prediction errors may be more influential.
We urge caution in interpreting results even on non terminal branches for which there are few sequences.
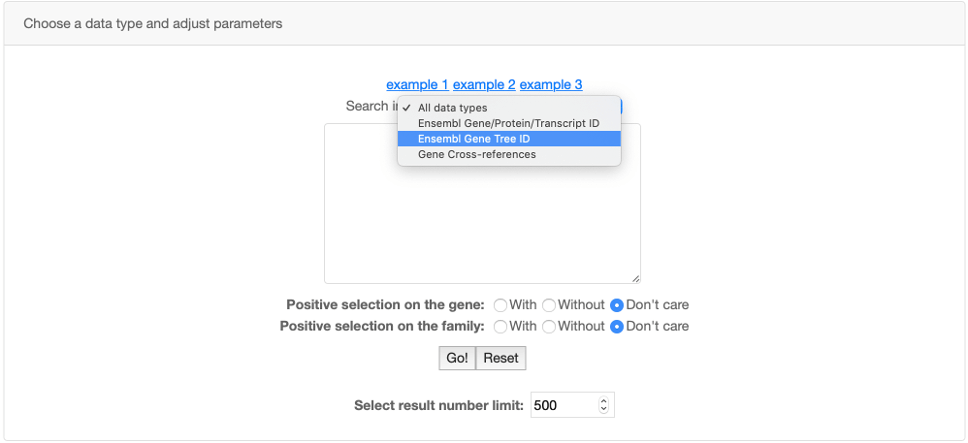
You can also use the Advanced search panel to restrict your search to a specific data type and also to search lists of Ensembl or Cross-References IDs.
After a Basic or an Advanced search, Selectome provides the results as a sortable table:
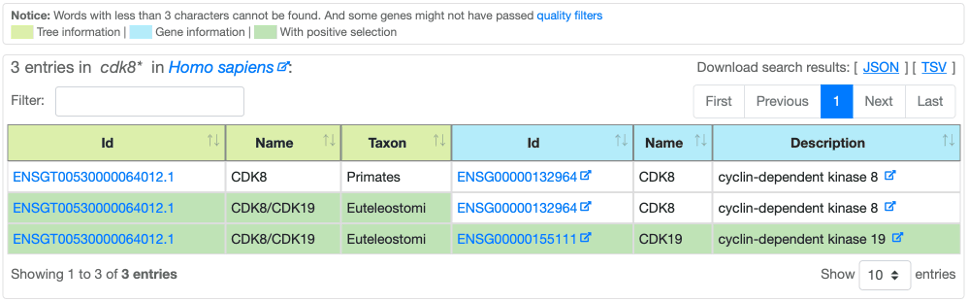
Leftmost columns are tree information.
Rightmost columns are gene information.
Background table cell highlights positive selection, if any, for the whole tree and/or per gene.
Table results can be exported in JSON or TSV formats.
Clicking on a gene family accession number shows the result of positive selection tests in more detail.
The family datasheet is split into two parts:
1. The description of the family, with accession number, symbol, and links to other sub-family members, if any.
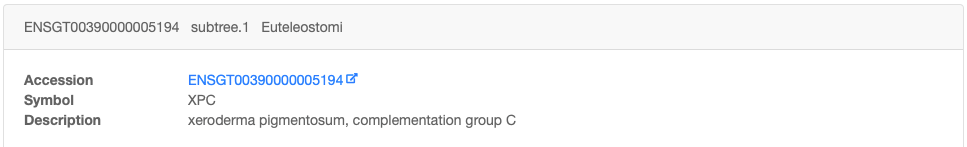
2. The subtree with added information on positive selection, but split - based on the taxon root used e.g. Euteleostomi - in sub-families.
You can click on gene names to access gene specific data.
Nodes appear as blue boxes for speciation events, as red for duplication events. Green boxes and branches are under positive selection.
Placing the mouse above a node provides information on:
- the type of event (speciation or duplication)
- the selection with p-value (if available; small p-values are more significant)
- the taxon
- the branch length
- the bootstrap (phylogenetic support for the branch, in %; high bootstrap values are more significant)
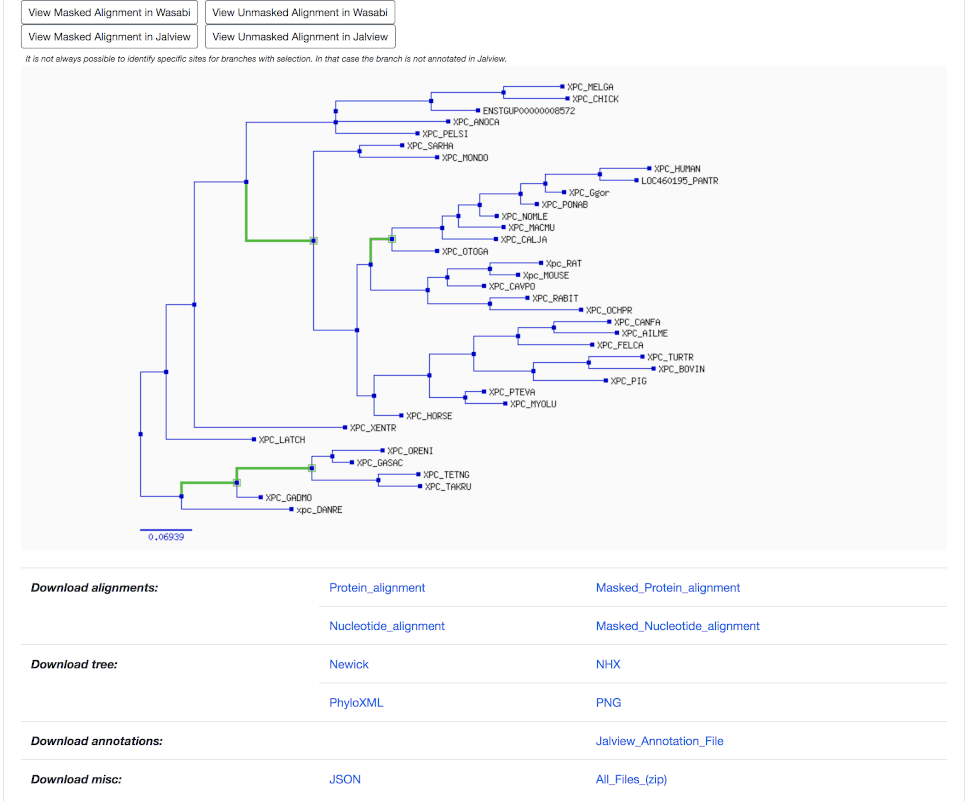
The last section provides links to download tree and alignment in various formats.
Finally, clicking on the View Masked Alignment in Wasabi button, or on the View Unmasked Alignment in Wasabi button, will display the protein alignment using the Wasabi tool.
Clicking on the View Masked Alignment in Jalview button, or on the View Unmasked Alignment in Jalview button, will display the protein alignment using the Jalview tool.
Masked alignment is the one out of our filtering pipeline, used for Godon computations. Unmasked alignment keeps original residues, before filtering.
Important note: Although the test on a branch may significantly detect positive selection over a relatively large proportion of sites (e.g. 5%), data will often not be sufficient to detect all these sites with good confidence. Thus in many cases fewer sites will be reported by BEB than expected from the likelihood test. And in some cases no sites will be predicted by BEB although positive selection is significant for a proportion of sites on the branch.
Wasabi
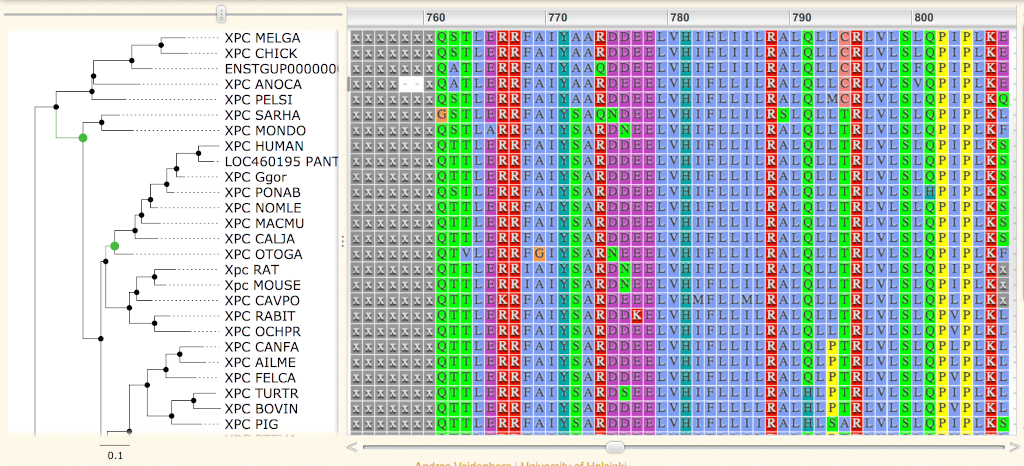
Jalview
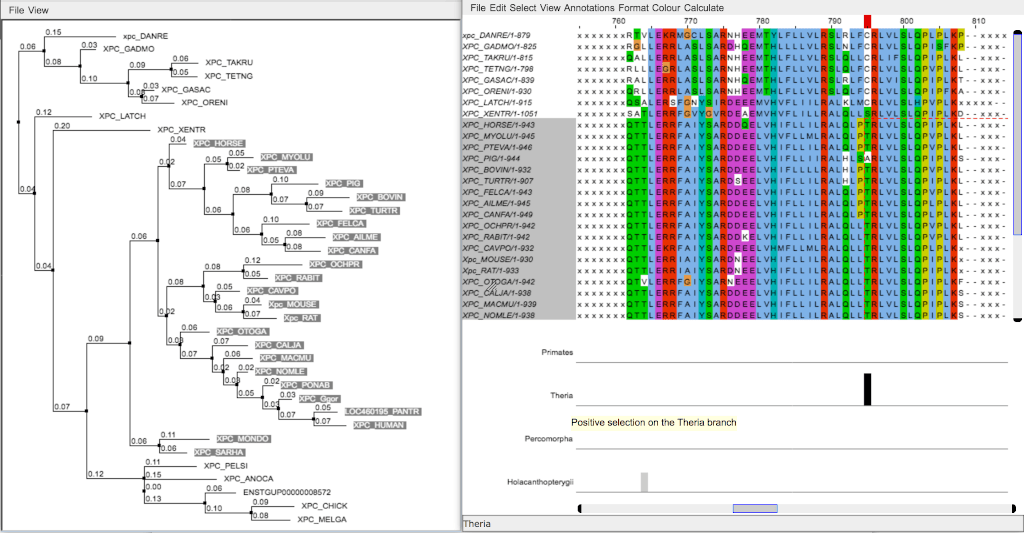
Default coloring follows the ClustalX code. Hydrophobic are in light blue (AVILWC), basic are in red (KR), acid are in purple (ED), polar are in green (NSTQ), aromatic polar in ocean (YH), proline in yellow (P), glycine in orange (G) and fully conserved cysteine in pink (C).
Masked residues are in lowercase x. Uppercase X are from the original sequence.